Demonstrating the integrity and security of laboratory data, records, results and
information is paramount for a successful audit or inspection for any GMP regulated
quality control laboratory.
According to FDA's Pre Approval Inspection Program 7346.832, the FDA Inspector
has to "audit the raw data, hardcopy or electronic, to authenticate the data submitted in the
CMC section of the application. And to verify that all relevant data (e.g. stability, biobatch data)
were submitted in the CMC section such that CDER product reviewers can rely on the
submitted data as complete and accurate".
In the compilation of a few selected Warning Letters issued in 2013 you will find the
key points regarding Laboratory Data Integrity.
The inspection focuses can be summarised and compiled in the following four themes:
1. Test Results / (Raw) Data
2. Written (Control) Procedures
3. Computerised Systems / Laboratory Instruments
4. Data Integrity Practices
Firms are responsible for the accuracy and integrity of all raw data generated in the
analytical labs of the facilities. Records should demonstrate that each batch was
tested and met the release specifications. These FDA Warning Letters indicate FDA's
continued focus on data integrity as a central concern of its international inspection program.
|

Consultoria especializada em GMP/BPF para indústrias voltadas a Ciências da Vida!
quinta-feira, 27 de fevereiro de 2014
GMP News: Laboratory Data Integrity in FDA Warning Letters 2013
terça-feira, 25 de fevereiro de 2014
F.D.A. Commissioner Pledges More Coordination with Indian Regulators - NYTimes.com
F.D.A. Commissioner Pledges More Coordination with Indian Regulators
By NEHA THIRANI BAGRI Sajjad Hussain/Agence France-Presse — Getty ImagesDr. Margaret A. Hamburg, right, commissioner of the U.S. Food and Drug Administration, with Anand Sharma, minister for Commerce and Industry, after a meeting in New Delhi on Feb. 10.
Sajjad Hussain/Agence France-Presse — Getty ImagesDr. Margaret A. Hamburg, right, commissioner of the U.S. Food and Drug Administration, with Anand Sharma, minister for Commerce and Industry, after a meeting in New Delhi on Feb. 10. MUMBAI — Dr. Margaret A. Hamburg, the commissioner of the United States Food and Drug Administration, said Tuesday that the American regulator would be working more closely with its Indian counterparts through training programs to ensure that Indian drug exporters meet American standards.
The announcement follows increased F.D.A. scrutiny of Indian drug makers over the past year, resulting in many warning letters to Indian companies, informing them that they did not meet the standards of the regulator. Dr. Hamburg has been in India for the past week meeting with the Indian regulators and government officials, policy makers and industry leaders who are involved in the regulation of medical and food products exported to the United States.
Dr. Hamburg announced at a news conference on Tuesday that the F.D.A would increase engagement with its Indian regulatory counterparts to better coordinate regulatory oversight, including holding seminars, webinars, meetings and training sessions.
“Training and capacity building was a theme heard over and over again from our counterpart regulatory authorities, industry and the research community,” said Dr. Hamburg. “We’re hoping to think about some new mechanisms of collaboration that will engage across industry, academia and the regulatory authorities in both of our countries.”
The F.D.A. inspected 160 Indian drug plants last year, which is three times the number inspected in 2009. Dr. Hamburg said the higher frequency of inspections and notices by the F.D.A.was not a uniquely Indian phenomenon, but was part of a global effort by the regulator to have tighter oversight on drugs produced overseas.
Responding to criticism from Indian regulators about overregulation she said, “We are not here to tell the Indian regulator how to do their job. But for companies that want to sell their products in the U.S. marketplace, then they do need to comply with our standards and practices and expectations, and we think that through greater collaboration we can enhance understanding about what our standards and expectations are.”
Dr. Hamburg also stressed the importance of Indian regulators engaging more deeply with international regulators.
“We work together as a coalition of international regulators to try to harmonize standards and approaches and share information, and often at those meetings India is not represented,” said Dr. Hamburg. “As India is such an important player on the global scene, we really hope that they will join us at the table.”
Last week, the F.D.A. signed a statement of intent with India’s health secretary, Keshav Desiraju, that said Indian inspectors would be allowed to observe firsthand the F.D.A.’s inspections of the manufacturing processes of pharmaceutical companies operating in India. As per the agreement, the Indian regulator would not be allowed to inform the company in advance of an inspection visit.
India’s Central Drug Standard Control Organization, the country’s drug regulator, has a staff of 323, about 2 percent the size of the F.D.A.’s.
Dr. Hamburg also said that because of the increased exports of Indian drugs to the United States, the American regulator is looking to increase its staff in India in the near future, from 12 to 19. The F.D.A. later said the latter figure included 10 inspectors dedicated specifically to medical products. She added that the F.D.A. has been making an effort to address the backlog in the approvals and pending applications. As of January, the F.D.A had cleared 45 percent of the backlog, she said.
India is one of the largest exporters of over-the-counter and prescription drugs to the United States, and its pharmaceutical industry supplies 40 percent of the over-the-counter and generic prescription drugs consumed in the United States.
“Indian drug manufacturers are extremely cost-efficient and feature among the top 10 generic drug companies worldwide,” said Sarabjit Kour Nangra, vice president of research for the pharmaceutical industry at Angel Broking in Mumbai. “So India is not a country the United States can ignore because of the economics of the business.”
Even taking into account the recent F.D.A. bans on certain Indian drugs, she said, exports to the United States by Indian drug makers continue to be robust.
Indian analysts said that most Indian pharmaceutical companies have been responsive to the F.D.A. inspections and that they are making an effort to tighten operations and meet global standards.
“Many Indian companies are trying to adapt and have invested in making sure that they reach U.S.F.D.A. standards,” said Ms. Nangra. “America is one of the largest geographies for Indian drug makers and one in which they have a big market share.”
However, she added that many Indian companies lacked people with expertise in world-class manufacturing and meeting the standards of the regulator.
Analysts said that increased stringency by the American regulator would lead to a significant increase in costs for Indian drug companies and make it harder for smaller pharmaceutical firms to enter the American market.
“The cost of doing business in the United States goes up because of compliance with F.D.A. norms to the extent that it makes it more difficult for the smaller players,” said Nitin Agarwal, director at IDFC Securities in India. “The United States is a difficult market to enter if you are a smaller firm and coming in late, but the focus on compliance now makes it a lot harder and more expensive.”
F.D.A. Commissioner Pledges More Coordination with Indian Regulators - NYTimes.com
Health Canada releases new GMP Guidelines for APIs
These Good Manufacturing Practices for Active Pharmaceutical Ingredients are designed to facilitate compliance by the regulated industry and to enhance consistency in the application of the regulatory requirements. It is also intended to help ensure that APIs meet the requirements for quality and purity that they purport or are represented to possess. The focus of these guidelines is on the manufacture of APIs sold in their final labeled container and/or used in the manufacture of finished dosage forms for human use. More specifically, they apply to the fabrication, packaging/labeling, testing, importation, distribution, wholesale, and re-packaging/re-labeling of APIs (including their intermediates).. The guide is available for download on the Health Canada website.
WEB source: Update on FDA and International GxP Compliance 2012
WEB source: Update on FDA and International GxP Compliance 2012
domingo, 23 de fevereiro de 2014
Remédio adia químio em câncer de próstata avançado - DIKAJOB
Resultados são de um estudo apresentado no Simpósio de Cânceres Geniturinários, em São Francisco
Fabiana Cambricoli, do 

Paciente com câncer recebendo tratamento de quimioterapia em um hospital de San Francisco
São Paulo - Um remédio para câncer de próstata atualmente indicado apenas a pacientes que passaram por quimioterapia se mostrou eficaz também no adiamento da necessidade do procedimento e na ampliação do tempo de sobrevida de homens com casos avançados da doença. Os resultados são de um estudo apresentado nesta quinta-feira, 30, no Simpósio de Cânceres Geniturinários, em São Francisco (Estados Unidos).
Na pesquisa, realizada pela Universidade de Saúde e Ciência de Oregon, 1.717 homens com tumor avançado de próstata foram divididos em dois grupos e acompanhados por 20 meses. Entre os que tomaram o medicamento enzalutamida, o risco de morte foi 29% inferior ao do grupo que usou um placebo. O tempo de sobrevida entre os que tomaram o remédio foi de 32,4 meses contra 30,2 meses dos pacientes que receberam o placebo.
De acordo com o estudo, o remédio ainda adiou em 17 meses o tempo médio de início da quimioterapia. De acordo com o chefe da equipe de urologia do Instituto do Câncer do Estado de São Paulo (Icesp), Rafael Coelho, participante do simpósio, o remédio será importante para prolongar a qualidade de vida dos pacientes com tumores avançados. "Ao retardar a necessidade de quimioterapia, o remédio adia também os efeitos colaterais da mesma", diz.
Coelho lamenta que a enzalutamida não esteja aprovada para uso no Brasil. "Sei que o processo de aprovação já foi aberto, mas por questões burocráticas, ainda não foi finalizado", diz. Nos EUA, a substância tem o aval desde 2012, mas apenas para uso depois da quimioterapia. Agora, após a apresentação do novo estudo, o fabricante pedirá à Administração de Alimentos e Medicamentos (FDA-Food and Drugs Administration), que a droga seja liberada também para uso antes da quimioterapia. O processo deve durar alguns meses.
Procurada pela reportagem, a Agência Nacional de Vigilância Sanitária (Anvisa) afirmou que o processo de aprovação da droga está em análise desde fevereiro e que, atualmente, aguarda informações pedidas ao fabricante para que sejam avaliadas a segurança e a eficácia do produto. Não há previsão para o término do processo. O tumor de próstata é o mais comum entre homens no Brasil, atrás apenas do câncer de pele não melanoma. De acordo com estimativas do Ministério da Saúde, 68,8 mil novos casos deverão ser registrados no País em 2014.
Fonte: Remédio adia químio em câncer de próstata avançado - DIKAJOB
Depyrogenation and Sterilization of Medical Device Materials with Nitrogen Dioxide Gas
Please read the article attached.
Depyprogenation and Sterilization with Nitrogem Dioxide Gas
Web source:http://www.noxilizer.com/
Depyprogenation and Sterilization with Nitrogem Dioxide Gas
Web source:http://www.noxilizer.com/
sábado, 22 de fevereiro de 2014
Interfarma - Frente parlamentar quer engajar Legislativo na redução de tributos sobre medicamentos
20/02/2014

Fotos: Mauricio de Souza
O presidente da Assembleia Legislativa, Samuel Moreira, reuniu-se nesta quinta-feira, 20/2, com a presidente da Frente Parlamentar para a Desoneração Tributária dos Medicamentos, deputada Maria Lúcia Amary (PSDB), e representantes da cadeia de produção, distribuição e venda de medicamentos. Eles pediram o apoio do Legislativo, junto ao governo do Estado, na obtenção da redução dos impostos incidentes sobre remédios.
Também foram entregues a Moreira cópias do primeiro e do último caderno (de número 3.824) do abaixo-assinado em favor da desoneração, que, disponível em cerca de 6 mil farmácias de todo o Estado, coletou em um mês 2.620.248 assinaturas e foi entregue, no último dia 12, aos presidentes da Câmara dos Deputados e do Senado.
"Queremos a ajuda do presidente da Assembleia e do líder do Governo para sensibilizar o governador quanto a essa proposta. São Paulo pode ser pioneiro na desoneração tributária dos remédios, que é uma medida de enorme alcance social", afirmou Maria Lúcia.
Moreira observou que desoneração não pode partir de uma iniciativa da Assembleia, por impedimentos legais. "Mas não há dúvida quanto ao mérito da luta que vocês estão empreendendo. E temos diversas possibilidades de atuação, como representantes da população", completou. Ao final da reunião, ficou decidido que, com o apoio do presidente do Legislativo e do líder do Governo, deputado Barros Munhoz (PSDB), se buscará agendar um encontro com o governador Geraldo Alckmin e com o secretário da Fazenda, Andrea Calabi.
Munhoz afirmou que existe disposição do governo em colaborar com a iniciativa e que o atual governo tem feito frequentes reduções de impostos, por entender que quem produz não pode ser prejudicado por uma elevada carga tributária. "Entendemos a justiça do que está sendo pleiteado e um dos principais papéis do parlamentar é exatamente encaminhar essas demandas ao Poder Executivo", destacou.
Representantes do setor farmacêutico deram respaldo às propostas discutidas na reunião. Segundo Pedro Bernardo, da Associação da Indústria Farmacêutica de Pesquisa (Interfarma), se o ICMS incidente sobre medicamentos fosse reduzido de 18% para 12%, não haveria perda para o Estado, já que os valores aí reduzidos seriam ganhos com o aumento da arrecadação em um dos elos da cadeia do setor: a distribuição de medicamentos.
"Quando a população toma conhecimento da carga de impostos sobre os medicamentos, a reação é de indignação. Acredito que a partir de agora ela não vai abrir de mão de conseguir [a redução], porque é uma questão de justiça social", acrescentou Bernardo.
Serafim Branco Neto, assessor da presidência da Associação Brasileira das Redes de Farmácias e Drogarias (Abrafarma), foi quem entregou ao presidente da Assembleia os cadernos do abaixo-assinado. "Nós, do varejo, sentimos o drama da população, quando ela vai em busca do medicamento", lembrou.
Também participaram da reunião os deputados Welson Gasparini (PSDB), Rafael Silva (PDT), Hélio Nishimoto (PSDB) e Chico Sardelli (PV). O deputado federal Valter Ihoshi (PSDB/SP) informou que após o carnaval deverá estar constituída uma comissão especial, no âmbito da Câmara Federal, para analisar as cercas de 20 propostas que tratam da questão e estão tramitando em Brasília. "A entrega do abaixo-assinado em Brasília representou uma grande vitória", ele concluiu.
Interfarma
quinta-feira, 20 de fevereiro de 2014
Brasil pode se tornar 4° mercado de medicamentos no mundo
Brasil pode se tornar 4° mercado de medicamentos no mundo
É curioso que, com um crescimento anual do Produto Interno Bruto (PIB) em torno de apenas 2,5%, ainda possamos deparar com situações típicas de economia aquecida, como, por exemplo, índices elevados de emprego, aumento de salário real, reajustes de preços dos serviços bem acima da média, importações crescentes, rodovias saturadas, filas de caminhões na entrada dos portos, inflação persistente etc. Alguns economistas diriam que temos uma insuficiência de oferta, e não de excesso de demanda.
O problema fica mais complexo quando os dados de cada grande segmento da economia são desagregados. Fiquei surpreso ao saber que a produção de sabonetes chegou a crescer 16% e o mercado de sorvetes mais de 15%. Ou que as atividades econômicas em municípios como Rondonópolis, no Mato Grosso, ou Três Lagoas, no Mato Grosso do Sul, expandem-se a um ritmo de 10% ao ano!
Esses descompassos setoriais, regionais ou locais talvez gerem uma dinâmica que explique o fato de, com um crescimento médio nada espetacular, vários indicadores se mostrarem conflitantes com o conjunto. Recentemente soube que faltam dois mil motoristas de ônibus no Rio de Janeiro (RJ). Sim, a cidade está com obras de infraestrutura por todos os lados (impulsionadas tanto pela preparação dos Jogos Olímpicos de 2016 como pelos investimentos que envolvem a cadeia produtiva da indústria do petróleo). Dirigir uma betoneira de concreto, um caminhão basculante ou um trator tem sido mais atrativo, profissionalmente, do que se estressar em vias engarrafadas, atendendo a uma clientela que reclama da qualidade do serviço oferecido.
Nesse ambiente, será um desafio contratar, e treinar em prazo curto, pessoal temporário para trabalhar em funções que direta ou indiretamente se relacionarão com a Copa do Mundo. E não se trata somente das localidades que servirão de sede para os jogos. Pelo número de inscrições de candidatos que se inscreveram pela internet para comprar ingressos na Copa, é quase certo que a previsão de que 600 mil turistas estrangeiros virão ao Brasil nas semanas dos jogos já esteja defasada. E os deslocamentos de uma cidade sede para outra movimentarão um milhão de brasileiros de diferentes Estados.
O futebol é, sem dúvida, atualmente o evento esportivo – e de entretenimento – que mais desperta interesse do público no planeta. Virão dos Estados Unidos, onde o que eles chamam de soccer tem feito mais sucesso entre as meninas, mais torcedores do que da vizinha Argentina. Na Espanha, em crise profunda, os estádios estão sempre cheios. As transmissões pela TV alcançam sempre índices elevados de audiência (é provável que o enorme sucesso da televisão por assinatura no Brasil, que dobrou sua base de expectadores, apenas em dois anos, tenha a ver também com os pacotes pay per view dos campeonatos nacional e estaduais).
Os efeitos da Copa sobre a economia brasileira não se limitam ao período dos jogos. Começaram antes, com obras de infraestrutura, e se estenderão por vários meses, em decorrência do turismo motivado pela exposição que o País terá durante o evento. Somados, esses efeitos poderiam corresponder a um acréscimo de 1,5 ponto percentual no nosso PIB. Nas projeções para 2014 talvez os economistas estejam subestimando esse impacto da Copa.
2014 será um ano de eleições gerais. O Governo fará grande esforço (inclusive por interesse eleitoral) para que as concessões de infraestrutura saiam do papel – aeroportos, portos, rodovias, ferrovias e hidrovias. Os governantes que pretendem se reeleger ou trabalham por seus aliados políticos têm até junho para cortar a “fita” de inauguração de obras. Empenhos (equivalente a autorizações para novos gastos públicos) relativos à contratação de obras não emergenciais não poderão ser feitos no segundo semestre.
Um crescimento da ordem de 3,5% em 2014 seria então plausível, apesar das taxas de juros mais salgadas que o Banco Central tem sido obrigado a adotar para que a inflação não saia de controle. Do câmbio não devem vir pressões inflacionárias expressivas (embora previsões sobre o comportamento de moeda costumem desmoralizar quem se arrisca a fazer prognósticos sobre elas), mas os combustíveis ficarão mais caros, assim como a energia elétrica. Em 2014, os governantes não vão querer o risco de postergar reajuste em tarifas de transporte. Os reajustes correrão até março ou ficarão represados, como bomba relógio, para 2015.
Saúde
Por causa de todas essas peculiaridades, algumas multinacionais do setor farmacêutico trabalham com a hipótese de o Brasil se tornar o quarto mercado de medicamentos no mundo, avançando pelo menos três casas no ranking internacional. E isso não teria relação somente com as compras governamentais (o SUS ultrapassou a Associação dos Veteranos de Guerra dos Estados Unidos como maior comprador isolado de remédios no mundo). A formalização dos empregos, a manutenção dos índices de ocupação em patamar elevado, acessos a planos corporativos e a prevenção de doenças relacionadas ao ritmo de vida nos centros urbanos e ao envelhecimento da população levam mais brasileiros a negligenciarem menos com sua própria saúde.
Há segmentos correlatos ao setor farmacêutico com perspectivas similares. É o caso dos fabricantes de produtos veterinários. O Brasil já é o maior exportador de carne bovina, disputa a liderança também na avicultura e está em quinto na suinocultura. Por força da demanda de alguns países vizinhos (Venezuela, especialmente), nos tornamos também exportadores de lácteos. Além de vacinas para prevenção de doenças e medicamentos para tratamento desses animais, há uma considerável produção de produtos veterinários para o mercado de pets. Estima-se em 50 milhões (35 milhões de cães e 15 milhões de gatos) essa população. O Brasil já é hoje o segundo mercado mundial de rações para pets!
Nos pontos de varejo (farmácias e drogarias) é crescente a participação de produtos de higiene pessoal e beleza. Explica-se: já somos o segundo mercado para xampus – recentemente uma grande multinacional francesa resolveu instalar um centro de pesquisa no Rio de Janeiro por causa da potencialidade do mercado brasileiro, e porque no País temos nada menos que oito dos nove tipos de cabelo encontrados no mundo, devido à imigração de diferentes etnias – e o quarto em cosméticos. O segundo ou terceiro perfume (uma água de Colônia) mais vendido no mundo é brasileiro, devido ao grande volume de vendas domésticas.
George Vidor é comentarista econômico da Globonews e colunista do jornal O Globo
Fonte: ICTQ
O problema fica mais complexo quando os dados de cada grande segmento da economia são desagregados. Fiquei surpreso ao saber que a produção de sabonetes chegou a crescer 16% e o mercado de sorvetes mais de 15%. Ou que as atividades econômicas em municípios como Rondonópolis, no Mato Grosso, ou Três Lagoas, no Mato Grosso do Sul, expandem-se a um ritmo de 10% ao ano!
Esses descompassos setoriais, regionais ou locais talvez gerem uma dinâmica que explique o fato de, com um crescimento médio nada espetacular, vários indicadores se mostrarem conflitantes com o conjunto. Recentemente soube que faltam dois mil motoristas de ônibus no Rio de Janeiro (RJ). Sim, a cidade está com obras de infraestrutura por todos os lados (impulsionadas tanto pela preparação dos Jogos Olímpicos de 2016 como pelos investimentos que envolvem a cadeia produtiva da indústria do petróleo). Dirigir uma betoneira de concreto, um caminhão basculante ou um trator tem sido mais atrativo, profissionalmente, do que se estressar em vias engarrafadas, atendendo a uma clientela que reclama da qualidade do serviço oferecido.
Nesse ambiente, será um desafio contratar, e treinar em prazo curto, pessoal temporário para trabalhar em funções que direta ou indiretamente se relacionarão com a Copa do Mundo. E não se trata somente das localidades que servirão de sede para os jogos. Pelo número de inscrições de candidatos que se inscreveram pela internet para comprar ingressos na Copa, é quase certo que a previsão de que 600 mil turistas estrangeiros virão ao Brasil nas semanas dos jogos já esteja defasada. E os deslocamentos de uma cidade sede para outra movimentarão um milhão de brasileiros de diferentes Estados.
O futebol é, sem dúvida, atualmente o evento esportivo – e de entretenimento – que mais desperta interesse do público no planeta. Virão dos Estados Unidos, onde o que eles chamam de soccer tem feito mais sucesso entre as meninas, mais torcedores do que da vizinha Argentina. Na Espanha, em crise profunda, os estádios estão sempre cheios. As transmissões pela TV alcançam sempre índices elevados de audiência (é provável que o enorme sucesso da televisão por assinatura no Brasil, que dobrou sua base de expectadores, apenas em dois anos, tenha a ver também com os pacotes pay per view dos campeonatos nacional e estaduais).
Os efeitos da Copa sobre a economia brasileira não se limitam ao período dos jogos. Começaram antes, com obras de infraestrutura, e se estenderão por vários meses, em decorrência do turismo motivado pela exposição que o País terá durante o evento. Somados, esses efeitos poderiam corresponder a um acréscimo de 1,5 ponto percentual no nosso PIB. Nas projeções para 2014 talvez os economistas estejam subestimando esse impacto da Copa.
2014 será um ano de eleições gerais. O Governo fará grande esforço (inclusive por interesse eleitoral) para que as concessões de infraestrutura saiam do papel – aeroportos, portos, rodovias, ferrovias e hidrovias. Os governantes que pretendem se reeleger ou trabalham por seus aliados políticos têm até junho para cortar a “fita” de inauguração de obras. Empenhos (equivalente a autorizações para novos gastos públicos) relativos à contratação de obras não emergenciais não poderão ser feitos no segundo semestre.
Um crescimento da ordem de 3,5% em 2014 seria então plausível, apesar das taxas de juros mais salgadas que o Banco Central tem sido obrigado a adotar para que a inflação não saia de controle. Do câmbio não devem vir pressões inflacionárias expressivas (embora previsões sobre o comportamento de moeda costumem desmoralizar quem se arrisca a fazer prognósticos sobre elas), mas os combustíveis ficarão mais caros, assim como a energia elétrica. Em 2014, os governantes não vão querer o risco de postergar reajuste em tarifas de transporte. Os reajustes correrão até março ou ficarão represados, como bomba relógio, para 2015.
Saúde
Por causa de todas essas peculiaridades, algumas multinacionais do setor farmacêutico trabalham com a hipótese de o Brasil se tornar o quarto mercado de medicamentos no mundo, avançando pelo menos três casas no ranking internacional. E isso não teria relação somente com as compras governamentais (o SUS ultrapassou a Associação dos Veteranos de Guerra dos Estados Unidos como maior comprador isolado de remédios no mundo). A formalização dos empregos, a manutenção dos índices de ocupação em patamar elevado, acessos a planos corporativos e a prevenção de doenças relacionadas ao ritmo de vida nos centros urbanos e ao envelhecimento da população levam mais brasileiros a negligenciarem menos com sua própria saúde.
Há segmentos correlatos ao setor farmacêutico com perspectivas similares. É o caso dos fabricantes de produtos veterinários. O Brasil já é o maior exportador de carne bovina, disputa a liderança também na avicultura e está em quinto na suinocultura. Por força da demanda de alguns países vizinhos (Venezuela, especialmente), nos tornamos também exportadores de lácteos. Além de vacinas para prevenção de doenças e medicamentos para tratamento desses animais, há uma considerável produção de produtos veterinários para o mercado de pets. Estima-se em 50 milhões (35 milhões de cães e 15 milhões de gatos) essa população. O Brasil já é hoje o segundo mercado mundial de rações para pets!
Nos pontos de varejo (farmácias e drogarias) é crescente a participação de produtos de higiene pessoal e beleza. Explica-se: já somos o segundo mercado para xampus – recentemente uma grande multinacional francesa resolveu instalar um centro de pesquisa no Rio de Janeiro por causa da potencialidade do mercado brasileiro, e porque no País temos nada menos que oito dos nove tipos de cabelo encontrados no mundo, devido à imigração de diferentes etnias – e o quarto em cosméticos. O segundo ou terceiro perfume (uma água de Colônia) mais vendido no mundo é brasileiro, devido ao grande volume de vendas domésticas.
George Vidor é comentarista econômico da Globonews e colunista do jornal O Globo
Fonte: ICTQ
Snif Brasil
FDA, dos EUA, aprova inovação israelense: uma pílula com câmera que ajuda no exame de pacientes com suspeita de câncer
Uma pílula com câmera para ajudar no exame de pacientes com suspeita de cãncer foi aprovada pela FDA (Food and Drug Administration), dos EUA, e em outros 80 países da Ásia, Europa e América Latina. A “PillCam” foi desenvolvida pela empresa israelense de tecnologia Given Imaging e pode ser usada em pacientes que têm dificuldades para realizar colonoscopia, um exame endoscópico do intestino que é realizado, principalmente, para detecção de pólipos (tumores benignos) que podem sofrer transformação para a malignidade.
A tecnologia, desenvolvida a partir de sistemas de defesa antimísseis, usa uma câmera portátil para tirar fotografias de alta velocidade durante a passagem pelo intestino ao longo de oito horas. As imagens são transmitidas para um dispositivo de gravação usado em torno da cintura do paciente e depois analisadas por um médico.
O sistema já existe desde 2001, mas não havia sido aprovado para substituir o exame tradicional, pois as imagens não eram tão claras. Por isso, o uso da pílula com a microcâmera era indicado apenas para pacientes que têm dificuldade para passar por colonoscopias convencionais. A cada ano, 750 mil pacientes não conseguem se submeter ao exame tradicional por problemas de anatomia ou cirurgias anteriores.
A pílula custa US$ 500, um valor bem mais baixo do que o exame tradicional, que normalmente sai por US$ 4.000. Os médicos também apostam no dispositivo para atrair pessoas que têm medo de sentir dor ou se sentem desconfortáveis com a colonoscopia tradicional.
Fonte:
Snif Brasil
A tecnologia, desenvolvida a partir de sistemas de defesa antimísseis, usa uma câmera portátil para tirar fotografias de alta velocidade durante a passagem pelo intestino ao longo de oito horas. As imagens são transmitidas para um dispositivo de gravação usado em torno da cintura do paciente e depois analisadas por um médico.
O sistema já existe desde 2001, mas não havia sido aprovado para substituir o exame tradicional, pois as imagens não eram tão claras. Por isso, o uso da pílula com a microcâmera era indicado apenas para pacientes que têm dificuldade para passar por colonoscopias convencionais. A cada ano, 750 mil pacientes não conseguem se submeter ao exame tradicional por problemas de anatomia ou cirurgias anteriores.
A pílula custa US$ 500, um valor bem mais baixo do que o exame tradicional, que normalmente sai por US$ 4.000. Os médicos também apostam no dispositivo para atrair pessoas que têm medo de sentir dor ou se sentem desconfortáveis com a colonoscopia tradicional.
Fonte:
Snif Brasil
FDA Updates Analytical Validation Guidance - Pharmaceutical Technology
FDA Updates Analytical Validation Guidance
FDA has released a draft guidance on analytical procedures and methods validation, which supersedes the 2000 draft guidance and will also replace the 1987 guidance, Submitting Samples and Analytical Data for Methods Validation. The draft guidance provides recommendations for submitting analytical procedures and methods validation data to support the documentation of the identity, strength, quality, purity, and potency of drug substances and drug products. The revised draft guidance complements International Conference on Harmonization (ICH) Q2(R1) Validation of Analytical Procedures: Text and Methodology.
The guidance is written to help with the assembly of information and presentation of data to support analytical methodologies. The recommendations apply to drug substances and drug products covered in new drug applications (NDAs), abbreviated new drug applications (ANDAs), biologics license applications (BLAs), and supplements to these applications. The principles in the revised draft guidance also apply to drug substances and drug products covered in Type II drug master files (DMFs). This revised draft guidance does not address investigational new drug application (IND) methods validation, but sponsors preparing INDs may want to consider the recommendations in the guidance.
According to the draft guidance, INDs require sufficient information at each phase of an investigation to ensure proper identity, quality, purity, strength, and/or potency. The amount of information on analytical procedures and methods validation will vary with the phase of the investigation. For general guidance on analytical procedures and methods validation information to be submitted for phase one studies, FDA recommends sponsors refer toGuidance for Industry, Content and Format of Investigational New Drug Applications (INDs) for Phase 1 Studies of Drugs, Including Well-Characterized, Therapeutic, Biotechnology-Derived Products. General considerations for analytical procedures and method validation (e.g., bioassay) before conduct of phase three studies are discussed in Guidance for Industry, IND Meetings for Human Drugs and Biologics, Chemistry, Manufacturing, and Controls Information.
The revised analytical validation draft guidance does not address specific method validation recommendations for biological and immunochemical assays for characterization and quality control of many drug substances and drug products. Some bioassays, for example, are based on animal challenge models, and immunogenicity assessments or other immunoassays have unique features that should be considered during development and validation. In addition, the need for revalidation of existing analytical methods may need to be considered when the manufacturing process changes during the product’s lifecycle.
Source: FDA.gov
FDA Updates Analytical Validation Guidance - Pharmaceutical Technology
segunda-feira, 17 de fevereiro de 2014
Frente arrecada 2,7 milhões de assinaturas pela redução de impostos sobre remédios - Câmara Notícias - Portal da Câmara dos Deputados
A Frente Parlamentar para a Desoneração dos Medicamentos entrega nesta tarde abaixo-assinado com 2,7 milhões de assinaturas pela redução dos impostos sobre remédios. A lista será entregue ao presidente da Câmara, Henrique Eduardo Alves, às 15 horas.
Coordenada pelo deputado Walter Ihoshi (PSD-SP), a frente tem trabalhado para diminuir a carga tributária desses produtos no Brasil que, segundo o deputado, atinge quase 34%, enquanto a média mundial é de 6%. “Essa iniciativa, feita em seis mil farmácias e drogarias, buscou aproximar ainda mais a população do debate sobre o assunto, com o intuito de sensibilizar as esferas dos executivos estaduais e federal a ajudar nesta causa benéfica para todos", ressaltou Ihoshi.
Já confirmaram presença o ministro-chefe da Secretaria da Micro e Pequena Empresa, Guilherme Afif Domingos, deputados, senadores e representantes das entidades farmacêuticas.
Exemplo do Paraná
Segundo Walter Ihoshi, a ideia é negociar e convencer os governadores a adotar, ainda neste ano, o exemplo do Paraná, onde os remédios estão desonerados desde 2008. "Os governantes têm medo de diminuir imposto. Eles acham que vão perder arrecadação. No Paraná o que aconteceu? Triplicou a arrecadação sobre os medicamentos. O setor conseguiu repassar essa carga tributária que foi desonerada para o preço ao consumidor final e o consumidor pode comprar o remédio que ele não tinha condições. Aumentou a arrecadação porque aumentou o consumo."
Segundo Walter Ihoshi, a ideia é negociar e convencer os governadores a adotar, ainda neste ano, o exemplo do Paraná, onde os remédios estão desonerados desde 2008. "Os governantes têm medo de diminuir imposto. Eles acham que vão perder arrecadação. No Paraná o que aconteceu? Triplicou a arrecadação sobre os medicamentos. O setor conseguiu repassar essa carga tributária que foi desonerada para o preço ao consumidor final e o consumidor pode comprar o remédio que ele não tinha condições. Aumentou a arrecadação porque aumentou o consumo."
Arquivo/ Gustavo Lima

Walter Ihoshi: metade da população não tem dinheiro para comprar remédio.
O deputado destacou ainda que 52% da população brasileira não conseguem comprar remédios, por causa dos preços altos. Segundo o deputado, as pessoas não se dão conta de que pagam 34% de imposto ao comprar um medicamento, muito acima da média mundial que é de apenas 6%. "34% de todo o preço praticado nas farmácias corresponde a imposto, a tributo. E nós sabemos que as pessoas mais pobres, segundo as pesquisas, são as que mais sofrem com a alta carga do imposto sobre os remédios. R$ 7 é a média do gastos das pessoas pobres no Brasil."
O deputado lembrou ainda que defendeu, em audiência pública realizada em agosto na Câmara, a redução do Imposto sobre Circulação de Mercadorias e Serviços (ICMS) sobre medicamentos. Em outra audiência, em outubro, representantes da indústria farmacêutica e da indústria de produtos hospitalares e odontológicos pediram a aprovação de propostas que diminuam os tributos que incidem sobre esses produtos.
Reportagem - Idhelene Macedo
Edição – Regina Céli Assumpção
Edição – Regina Céli Assumpção
Frente arrecada 2,7 milhões de assinaturas pela redução de impostos sobre remédios - Câmara Notícias - Portal da Câmara dos Deputados
quinta-feira, 13 de fevereiro de 2014
GMP News: Revision of the EU GMP Annex 15 for Qualification and Validation published
n our news EMA published a concept paper on the revision of the Annex 15 we reported about the planned revision of Annex 15. Now, the European Commission has published the draft of the Annex 15.
Compared to the original 11 pages of Annex 15 the draft has now 17 pages. The above-mentioned concept paper is mentioned as a reason for the revision, i.e. adjustments to changes partially to part 1 of the EU GMP Guide, changes in Annex 11, adjustments to the ICH Q8-11 documents, to the EMA Process Validation draft and changes in manufacturing technologies.
Below is a short summary of the major changes.
The revision is very extensive. Influences of the ICH Guidelines Q8, 9, 10 (and 11) are clearly visible - even in the glossary. The subject Design Space (ICH Q8) is now also covered in the area Process Validation. Many risk considerations (ICH Q9) are now mandatory. The life cycle approach and process knowledge (ICH Q10) are now included as well. Deviation management gained in significance. Third party services are now authorized explicitly if the supplier has been qualified correspondingly. This is a positive adaptation to reality. The mention of preliminary approvals - e.g. in the case of deviations - is good for a next (validation / qualification) stage, if there is a documented assessment showing that there is no significant impact on the next stage. Unfortunately a clear differentiation between qualification (based on equipment and facilities) and validation (related to processes) is missing. This is a shortcoming that unfortunately exists in many European regulations.
Other important changes compared to the current version are:
Compared to the original 11 pages of Annex 15 the draft has now 17 pages. The above-mentioned concept paper is mentioned as a reason for the revision, i.e. adjustments to changes partially to part 1 of the EU GMP Guide, changes in Annex 11, adjustments to the ICH Q8-11 documents, to the EMA Process Validation draft and changes in manufacturing technologies.
Below is a short summary of the major changes.
The revision is very extensive. Influences of the ICH Guidelines Q8, 9, 10 (and 11) are clearly visible - even in the glossary. The subject Design Space (ICH Q8) is now also covered in the area Process Validation. Many risk considerations (ICH Q9) are now mandatory. The life cycle approach and process knowledge (ICH Q10) are now included as well. Deviation management gained in significance. Third party services are now authorized explicitly if the supplier has been qualified correspondingly. This is a positive adaptation to reality. The mention of preliminary approvals - e.g. in the case of deviations - is good for a next (validation / qualification) stage, if there is a documented assessment showing that there is no significant impact on the next stage. Unfortunately a clear differentiation between qualification (based on equipment and facilities) and validation (related to processes) is missing. This is a shortcoming that unfortunately exists in many European regulations.
Other important changes compared to the current version are:
- There are new terms that are not explained in the glossary, such as "ongoing validation strategy"
- Retrospective validation and the notion of revalidation are gone completely - except for one exemption
- The qualification has become more extensive due to the integration of user requirements as a separate step and claim for a SAT.
- Information regarding the qualification of equipment in use have been omitted completely.
- The naming of transportation verification, packaging validation, validation of utilities and validation of analytical methods are completely new.
- In the Process Validation there are now 2 different approaches - a (modern) "continuous verification" approach and a traditional approach. The latter is still based on the classic 3 validation runs. But in any case, process robustness has to be proven.
- "Bracketing" approaches in the Process Validation are possible now - have to be justified though
- A hybrid approach between continuous verification and the traditional Process Validation approach remains somewhat "fuzzy"
- With regard to cleaning validation the 'visibly clean' criterion as the only acceptance criterion is no more acceptable.
- A grouping of equipment in the cleaning validation is explicitly possible now - but needs to be founded
- Acceptance criteria for a cleaning validation depend on toxicological data (permitted daily exposure, PDE)
- The statement that the number of cleaning validation runs has to be decided on the basis of a risk assessment is very interesting. The magic 3 is not mentioned explicitly here - unlike the Process Validation.
- The concept of cleaning verification for rarely manufactured products is also new
- The demand for "dirty and clean-hold times" is an adaptation to the state of the art
Conclusion: All in all there is an abundance of new requirements, which partly only represent the state of the current technology. And partly the "problems" remain in the details. A detailed analysis will follow.
The draft comment period is relatively short and ends (unfortunately) already end of May 2014.
The draft comment period is relatively short and ends (unfortunately) already end of May 2014.
Here you will find the Draft EU GMP Annex 15 for Download
GMP News: Revision of the EU GMP Annex 15 for Qualification and Validation published
GMP News: 85 Company listed with GMP-Non Compliance Statements in the New EMA Database
As already reported, the EMA has started listing so-called GMP-Non-Compliance Statements in the EudraGMDP database in addition to Certificates of GMP Compliance. In future, a similar procedure should apply to certificates of GDP Non-Compliance.
All European GMP and GDP inspection authorities enter the results of inspections in this database. Certificates of GMP Compliance have been extensively entered and updated, now it's time for Non-Compliance Reports to be published. Currently, there are 85 GMP Non-Compliance Statements in the EMA database. The results are both as clear as notable: companies from India and China are - by far - the most represented. The diagram is showing the countries where GMP non-compliant facilities have been identified. One may question how many facilities in China and India do not manufacture under GMP. Not only the number of GMP non-compliant facilities is frightening but also the GMP deviations themselves (details will follow). Beside insufficient GMP implementation, the falsification of data has been observed several times. Based on those data, manufacturers of medicinal products who import APIs from India and China have to check whether their own GMP audits have been sufficiently performed. The situation is also critical for the Qualified Person. Should a medicinal product cause any risk to patient safety (in extreme cases even death) because of the GMP non-compliant production of an API, a court would probably verify how accurate and to what extent facilities in the Far East have been monitored. Some companies might have difficulties to prove the complete and gapless monitoring of their suppliers. At this point - if not before - several so-called "one day audits" should be critically questioned.
All European GMP and GDP inspection authorities enter the results of inspections in this database. Certificates of GMP Compliance have been extensively entered and updated, now it's time for Non-Compliance Reports to be published. Currently, there are 85 GMP Non-Compliance Statements in the EMA database. The results are both as clear as notable: companies from India and China are - by far - the most represented. The diagram is showing the countries where GMP non-compliant facilities have been identified. One may question how many facilities in China and India do not manufacture under GMP. Not only the number of GMP non-compliant facilities is frightening but also the GMP deviations themselves (details will follow). Beside insufficient GMP implementation, the falsification of data has been observed several times. Based on those data, manufacturers of medicinal products who import APIs from India and China have to check whether their own GMP audits have been sufficiently performed. The situation is also critical for the Qualified Person. Should a medicinal product cause any risk to patient safety (in extreme cases even death) because of the GMP non-compliant production of an API, a court would probably verify how accurate and to what extent facilities in the Far East have been monitored. Some companies might have difficulties to prove the complete and gapless monitoring of their suppliers. At this point - if not before - several so-called "one day audits" should be critically questioned.
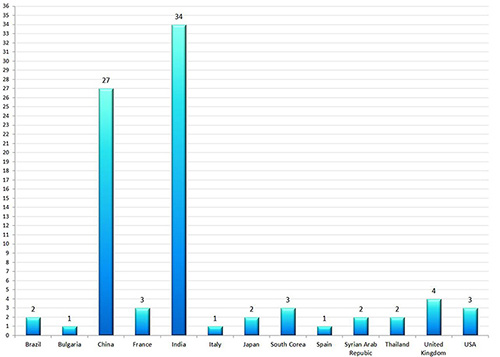
The EMA goes even far beyond the FDA; whereas FDA's Warning Letters represent (often) a last chance for companies to get rid of deviations, the Statement of Non Compliance means that the products in question are no longer marketable in the EU. So far, this information hadn't been available to the public.
Moreover, EMA's data disclose for each facility concerned the respective GMP deviation. Below please find some information about recent inspection findings: in October 2013, the French authority performed an inspection at Smruthi Organics Limited in India. There, considerable deviations were observed and the authority issued a GMP Non-Compliance Statement in the EudraGMDP. If we have a look at the deviations, there were 29 in total - among them 2 critical and 4 major ones. Critical deviation 1 concerned the manipulation and falsification of documents and data and was found in different departments. Critical deviation 2 was about corrective and preventive measures with regard to deviations identified during a past inspection which haven't been processed satisfactorily and removed.
As a consequence of the inspection performed within the EDQM inspection programme, 6 CEPs have been withdrawn. In addition, it is pointed out that another inspection already performed on 6 February 2013 had led to a Non-Compliance Statement for the company.
Besides, an API manufacturer (Suzhou No. 4 Pharmaceutical Factory in China) also got a GMP Non-Compliance Statement - in that case delivered by the Norwegian authority. The inspection outcome revealed 3 critical and 4 major deviations from the GMP regulations. The critical deviations were identified in the areas of Quality Management and Cleaning of Facilities. The major GMP deviations referred to Premises, Quality Control, and Change Control. There had already been a Non GMP Compliance Statement for this company too after an earlier inspection. After the second inspection, the EDQM decided to withdraw 3 CEPs.
Now that the EudraGMDP has been extended, it has become an essential element for the Compliance examination of facilities. It enables manufacturers of medicinal products to rapidly determine whether EU authorities have issued GMP Non Compliance for an API manufacturer.
Moreover, EMA's data disclose for each facility concerned the respective GMP deviation. Below please find some information about recent inspection findings: in October 2013, the French authority performed an inspection at Smruthi Organics Limited in India. There, considerable deviations were observed and the authority issued a GMP Non-Compliance Statement in the EudraGMDP. If we have a look at the deviations, there were 29 in total - among them 2 critical and 4 major ones. Critical deviation 1 concerned the manipulation and falsification of documents and data and was found in different departments. Critical deviation 2 was about corrective and preventive measures with regard to deviations identified during a past inspection which haven't been processed satisfactorily and removed.
As a consequence of the inspection performed within the EDQM inspection programme, 6 CEPs have been withdrawn. In addition, it is pointed out that another inspection already performed on 6 February 2013 had led to a Non-Compliance Statement for the company.
Besides, an API manufacturer (Suzhou No. 4 Pharmaceutical Factory in China) also got a GMP Non-Compliance Statement - in that case delivered by the Norwegian authority. The inspection outcome revealed 3 critical and 4 major deviations from the GMP regulations. The critical deviations were identified in the areas of Quality Management and Cleaning of Facilities. The major GMP deviations referred to Premises, Quality Control, and Change Control. There had already been a Non GMP Compliance Statement for this company too after an earlier inspection. After the second inspection, the EDQM decided to withdraw 3 CEPs.
Now that the EudraGMDP has been extended, it has become an essential element for the Compliance examination of facilities. It enables manufacturers of medicinal products to rapidly determine whether EU authorities have issued GMP Non Compliance for an API manufacturer.
Source: EudraGMDP Database
source: GMP News: 85 Company listed with GMP-Non Compliance Statements in the New EMA Database
Assinar:
Postagens (Atom)